
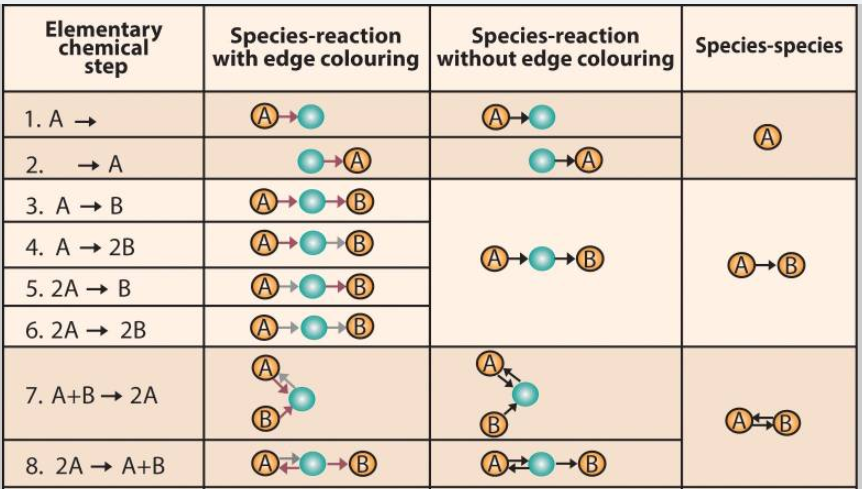
Systems chemists increasingly use network representation to investigate chemical pathways and their group. With the advent of high-performance scientific computing, graph databases (like Neo4j) gradually gain its popularity in representing chemical interaction. I will introduce how the network model and graph database boost the process to find a candidate for a particular experiment. As demonstrated in the graph below, a complex reaction mechanism can be represented by a set of elementary chemical reactions that are easily translated into a directed graph model. In this case, one node represents either one reactant or product. For one reactant, it can point to its product with edges containing the information of the chemical reaction (like the rate of reaction, the speed of reaction, reaction condition, etc). If we can get a path from the reactant node to the required product node, then there is a series of chemical interactions that can transform the reactant into the product. Suppose a chemist wants to find the fastest way to get a chemical product (Denote as D) from a given reactant (Denote as A) in such a graph database. It is logically equivalent to find a path from node A to node D what has the largest product(?) of all reaction rate (denote as ki) along the path. A chemist can even choose a reaction path with the safest experiment condition if he filters the reaction condition stored on the edges (Denote as the relationship in a graph database). The chemical interaction database saves a huge amount of time for chemists to search for a potential pathway between reactant and product.
The graph representation also brings convenience to biochemists. The systems biology definition of a network is broader and includes a variety of graphs. The nodes in a network generally represent biochemical components. Some examples include genes and proteins in a transcription network; substrates, organic compounds and so on. For this type of graphs, one Strongly Connected Component (SCC) usually represents a stable group of components that can transform into each other under some conditions. Like the Bow-tie Structure taught in the first lecture, there is a similar structure in the graph representation of biochemical interactions. The components in SCC can be formed from the components that are in the IN set of it. Additionally, the SCC can produce the components that are in the OUT set of it. By using graph representation, biochemists can have a DAG of biochemical interaction where each SCC can be identified as a compound species. They can produce undiscovered biochemist compounds by permuting different SCCs. Therefore, nowadays scientists are computationally generating a large space of possible biophysical-chemical realistic pathways and then testing them for their potential to exhibit particular biochemical functions.
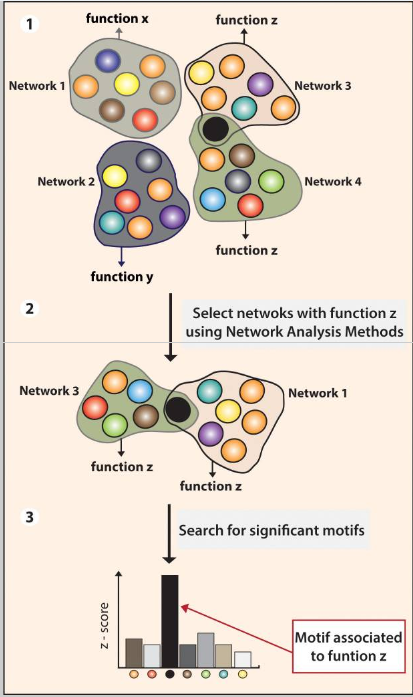
One SCC is denoted as one reaction network in biochemistry
During the past decades, network theory has been proposed to analyze chemical reaction systems and biochemical pathways. An essential question for translating chemical and biochemical reactions into a network model is whether the representation is accurate and whether any information is lost. With the development of modern graph database, modern chemists save a huge amount of time to build and search for a large-scale network representation of chemical and biochemical interactions.
Reference List:
[1] Sandefur, C. I., Mincheva, M., & Schnell, S. (2013). Network representations and methods for the analysis of chemical and biochemical pathways. Molecular BioSystems, 9(9), 2189. doi: 10.1039/c3mb70052f
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3755892/
[2] Kim, Y., Kim, J. W., Kim, Z., & Kim, W. Y. (2018). Efficient prediction of reaction paths through molecular graph and reaction network analysis. Chemical Science, 9(4), 825–835. doi: 10.1039/c7sc03628k
https://pubs.rsc.org/en/content/articlelanding/2018/sc/c7sc03628k#!divAbstract
[3] Organic Chemistry. (n.d.). Retrieved from:









{kind=link}